大家好,我是邓飞。
今天星球的小伙伴问了一个问题:
我现在在做GWAS分析,现在已经找到性状关联的SNP位点,下一步我如何根据position 找到基因呢?
关于基因注释,之前写过一些博客,可以用到的软件有:ANNOVAR、Bedtools,今天回答了这个问题,感觉excel也可以做基因注释了。
下面,对我的回答进行进一步的阐述。
1. GWAS分析
GWAS分析,之前写过一个Cookbook,包括方方面面的内容了,如果是小白,推荐一遍看配套的视频,一遍敲代码学习:
录制了配套的视频教程,前面的数据下载、软件安装、环境配置等相关视频免费观看,后面的付费观看。对于想要快速学习的小白,视频+代码+数据+实操+技术支持,是比较快的一条路。

(扫码查看视频教程)
2,显著SNP位点
做完GWAS分析后,确定阈值,然后小于阈值的位点都是显著性位点,显著性位点最重要的两个信息:
-
染色体
-
物理位置
有时候还包括snp的名称,但是不是必填项,只需要上面两个信息,就可以知道显著snp在基因组上的位置了。
3,配套基因组的gff文件
一般,有基因组数据的物种,有基因组的版本,还有配套的gff或者gff3格式的文件,文件的内容里面有:
-
染色体
-
基因起始位置
-
基因终止位置
-
基因功能描述
-
……
类似:
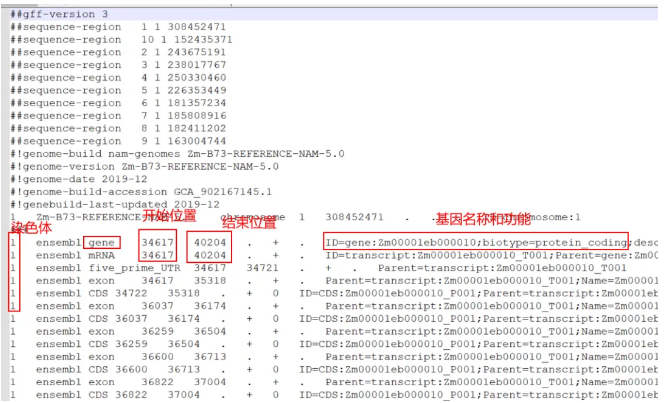
4,计算LD衰减距离
为何要计算LD衰减距离呢,是为了知道显著snp代表的区间,因为存在连锁,所以衰减距离就是确定snp所代表的有效区间,可以代表这个有效区间的变异。虽然snp不在基因上,但是如果snp的衰减距离区间内(比如上下50kb)包含基因,那也可以说明这个基因是显著影响性状的。
所以,计算了LD衰减距离,显著性snp的信息,就变成了:
-
染色体
-
有效区间起始位置
-
有效区间终止位置
5,用excel注释显著性snp
我们把gff文件,简化一下,整理成excel格式:
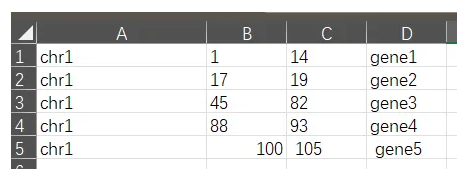
怎么用excel表格呢,可以手动查看,也可以编写一个函数。
话说,上面的显著性位点,一共就6个SNP,手动搞就行了。
第一个snp,区间是1染色体,5-15,这个区间有:gene1
第二个snp,区间是1染色体,10-20,这个区间有:gene2,不是完全包括,但是有交集,也算是
第三个snp,没有基因
第四个snp,gene4
第五个snp:没有基因
第六个snp:没有基因
所以这些snp,一共注释的基因有:gene1, gene2, gene4
6,我有1000个显著性位点,谢谢
如果位点很多,这就需要用到软件了:bedttols
「换到基因注释的领域,看一下相关需求:」
-
1,显著性的SNP位点,取上下游50k的位点,作为候选的区间
-
2,将候选区间有基因的,匹配到SNP的右边
「处理注意:」
-
1,显著SNP在上下游区间时,可能会有交叉,所以要先合并(merge)
-
2,匹配基因时,一个SNP区间可能会有多个基因
1. 数据描述
「SNP区间文件:」
这里,提取显著SNP的区间,提取三列信息:染色体,开始位置,结束位置:
共有6个SNP区间,其中第一个和第二个有重合,第五个和第六个有重合。
cat snp_infor.ped chr1 5 15 chr1 10 20 chr1 30 40 chr1 80 90 chr1 110 120「基因区间文件:」
共有5个基因区间文件,分别是:染色体,开始位置,终止位置,基因名称。
cat gene_infor.ped chr1 1 14 gene1 chr1 17 19 gene2 chr1 45 82 gene3 chr1 88 93 gene42. 提取每个SNP上面的基因
「需求:」
-
每个SNP一行
-
如果有基因在其区间,放到右边,如果没有基因,返回空
-
如果一个SNP区间对应多个基因,写成多行
代码:
-
intersect,交集
-
-a,第一个位置信息表
-
-b,第二个位置信息表
-
-loj,以第一个为基准,返回结果
结果可以看到,第二个SNP区间,对应两个基因,写成了两行。第三个SNP区间没有对应基因,用-1表示占位。共返回8行信息。
3. 返回有基因信息的SNP
如果不想要占位符,只想返回有基因的SNP信息,可以命令如下:
bedtools intersect -a snp_infor.ped -b gene_infor.ped -wa -wb
结果:
$ bedtools intersect -a snp_infor.ped -b gene_infor.ped -wa -wb chr1 5 15 chr1 1 14 gene1 chr1 10 20 chr1 1 14 gene1 chr1 10 20 chr1 17 19 gene2 chr1 80 90 chr1 45 82 gene3可以看到,将没有匹配到基因的SNP删除了。
上面的信息中,有些SNP匹配到了多个基因,也就是基因是有重复的。
-
如果我们想看每个SNP匹配的基因情况,可以用上面的结果
-
如果我们想看一下共有多少无重复的基因匹配,就需要对SNP区间先合并
4. 合并SNP区间再匹配
合并命令:
bedtools merge -i snp_infor.ped >snp_infor_merge.ped
原始数据:
$ cat snp_infor.ped chr1 5 15 chr1 10 20 chr1 30 40 chr1 80 90 chr1 110 120合并的结果:
$ cat snp_infor_merge.ped chr1 5 20 chr1 30 40 chr1 80 90然后和基因的信息进行合并:
$ bedtools intersect -a snp_infor_merge.ped -b gene_infor.ped -wa -wb chr1 5 20 chr1 1 14 gene1 chr1 5 20 chr1 17 19 gene2 chr1 80 90 chr1 45 82 gene35. 查看每个SNP区间基因的个数
结果可以用2中,统计一下个数,也可以用bedtools的-c参数:
$ bedtools intersect -a snp_infor.ped -b gene_infor.ped -c chr1 5 15 1 chr1 10 20 2 chr1 30 40 0 chr1 80 90 2 chr1 110 120 0结果可以看到,SNP1有一个基因,SNP2有2个基因,SNP3没有基因……
6. 基因注释的不同玩法
把上面SNP的区间,作为显著性SNP上下游的信息,把基因的信息作为gff基因文件,就可以进行基因注释了!
上面的玩法都可以做。
「注意,将gff格式整理为:染色体,开始位置,结束位置,基因信息;
snp区间整理为:染色体,开始区间,结束区间」
可以实现的功能:
-
每个SNP区间内的基因
-
每个SNP全进内基因的个数
-
合并SNP区间内的基因
-
合并SNP区间内基因的个数
本站资源均来自互联网,仅供研究学习,禁止违法使用和商用,产生法律纠纷本站概不负责!如果侵犯了您的权益请与我们联系!
转载请注明出处: 免费源码网-免费的源码资源网站 » GWAS分析中显著位点如何注释基因:excel???

发表评论 取消回复