pdb_strand_id:是历史上由 PDB 数据库定义的链标识符,用于区分同一PDB文件中不同的链(可以是多肽链或核酸链)。这是结构解析过程中最常见的链标识符。
asym_id:是 mmCIF 格式中使用的一个标识符,表示非对称单元的标识符。它通常表示结构中的一个独立的多肽或核酸链,并用于细致描述每个非对称单元的内容。
mmseqs2进行pdb蛋白质序列聚类分析结果中(mmseqs2进行pdb蛋白质序列聚类分析_mmseq2-CSDN博客),链的ID为pdb_strand_id,而 解析批量下载的mmCif复合体结构并保存为.pt格式文件(解析批量下载的mmCif复合体结构并保存为.pt格式文件-CSDN博客)结果中链的编号为asym_id。再下游模型训练时需要把cluster.csv(mmseqs2蛋白质聚类数据格式转化-CSDN博客) 中 pdb_strand_id转为asym_id。
示例代码:
### 蛋白质链的映射:pdb_strand_id 转 asym_id
import tempfile
from Bio.PDB import *
from mmcif.io.PdbxReader import PdbxReader
import os
def find_file_in_directory(file_name, search_directory):
# 遍历指定目录及其子目录
for root, dirs, files in os.walk(search_directory):
# 检查当前目录中的文件是否与指定文件名匹配
if file_name in files:
# 返回文件的完整路径
return os.path.join(root, file_name)
# 如果没有找到,返回None
return None
def parse_pdb_chain_to_asym_map(pdb_id, search_directory):
"""
biopython下载.cif结构文件到临时文件夹并解析链 PDB ID 到 asym ID 的映射
:param pdb_id: 目标蛋白质结构的 PDB ID
:return: 链 PDB ID 到 asym ID 的映射字典
"""
file_name = pdb_id.lower() + '.cif'
# 先查找有没有本地文件
file_path = find_file_in_directory(file_name, search_directory)
data = [] # 存储解析后的数据
if file_path:
with open(file_path, 'r') as file:
reader = PdbxReader(file)
reader.read(data)
else:
# 创建临时目录
with tempfile.TemporaryDirectory() as temp_dir:
pdbl = PDBList()
# 它将从服务器下载指定的文件(2fat.cif)并将其存储在当前工作目录中。
pdbl.retrieve_pdb_file(pdb_id, pdir = temp_dir, file_format = 'mmCif')
temp_file = os.path.join(temp_dir,file_name)
print(temp_file)
# 读取并解析 CIF 文件
with open(temp_file, 'r') as file:
reader = PdbxReader(file)
reader.read(data)
# 获取 pdbx_poly_seq_scheme 对象
block = data[0]
pdbx_poly_seq_scheme = block.getObj('pdbx_poly_seq_scheme')
# 提取 pdb_strand_id 和 asym_id_idx 的列索引
strand_id_idx = pdbx_poly_seq_scheme.getAttributeIndex('pdb_strand_id') # pdb_strand_id
asym_id_idx = pdbx_poly_seq_scheme.getAttributeIndex('asym_id') # asym_id
# 获取对应的值
strand_ids = pdbx_poly_seq_scheme.getColumn(strand_id_idx)
#print(pdb_strand_ids)
asym_ids = pdbx_poly_seq_scheme.getColumn(asym_id_idx)
# 加上pdb_id号
pdb_strand_ids = [f"{pdb_id}_{strand_id}" for strand_id in strand_ids]
pdb_asym_ids = [f"{pdb_id}_{asym_id}" for asym_id in asym_ids]
id_map = {}
for pdb_strand_id, pdb_asym_id in zip(pdb_strand_ids, pdb_asym_ids):
id_map.update([(pdb_strand_id, pdb_asym_id)])
return id_map
# 示例用法
pdb_id = "5d0j"
id_map = parse_pdb_chain_to_asym_map(pdb_id, "/Users/zhengxueming/test/mmcif")
print(id_map)批量转换并保存:
import csv
## 读取原始数据
with open("test_cluster.csv", 'r') as csvfile:
reader = list(csv.reader(csvfile))
data = reader[1:] # 读取数据部分
# 得到所有映射的字典
cluster_ids = [ item[1].split('_')[0] for item in data ]
homolog_ids = [ item[2].split(',') for item in data ]
homolog_ids = [ id.split('_')[0] for item in homolog_ids for id in item ]
#print(cluster_ids)
#print(homolog_ids)
pdb_ids = cluster_ids + homolog_ids
pdb_ids = set(pdb_ids)
#print(pdb_ids)
pdb_chain_to_asym_map = {}
for id in pdb_ids:
#print(id)
id_map = parse_pdb_chain_to_asym_map(id, "/Users/zhengxueming/test/mmcif")
pdb_chain_to_asym_map.update(id_map)
print(pdb_chain_to_asym_map)
## 根据new_chain_to_entity_map,替换test_cluster.csv中链PDB ID 为 entity ID并保存到新的csv文件中
with open("test_cluster.csv", 'r') as csvfile:
reader = list(csv.reader(csvfile))
data = reader[1:] # 读取数据部分
with open("new_test_cluster.csv", 'w', newline='') as csvfile:
writer = csv.writer(csvfile)
writer.writerow(["IDX","HOMOLOG"])
for idx, cluster_id, homolog_ids in data:
homolog_id_lst = homolog_ids.split(',')
all_id_lst = [cluster_id] + homolog_id_lst
writer.writerow([idx, ','.join([pdb_chain_to_asym_map[pdb_id] for pdb_id in all_id_lst if pdb_id in pdb_chain_to_asym_map.keys()])])
转化前:
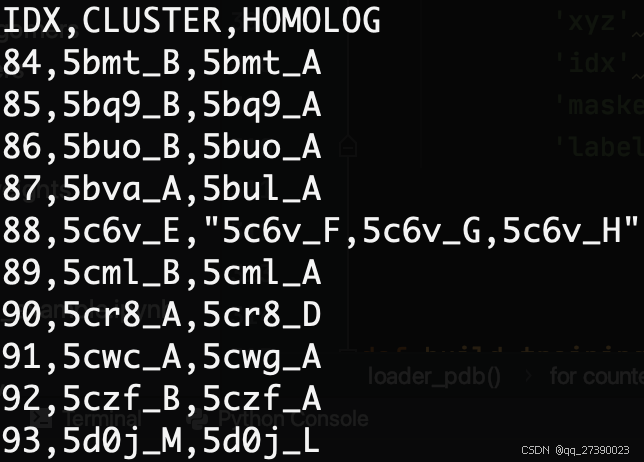
转化后:
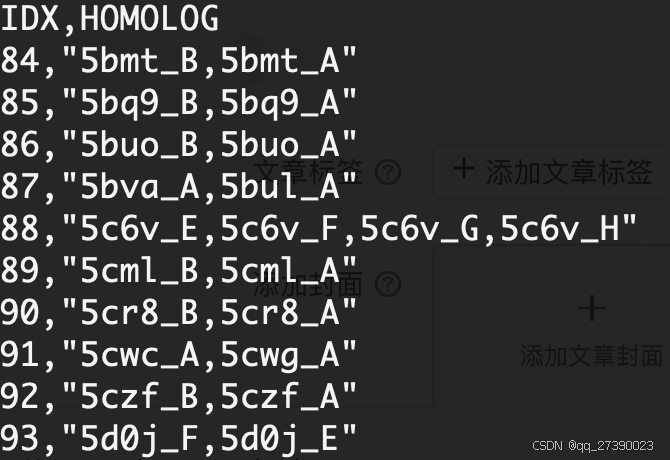
## 第一列为 index, 第二列为同源序列,其中第一个为代表序列(合并了原来的第二列和第三列后再转化)。
本站资源均来自互联网,仅供研究学习,禁止违法使用和商用,产生法律纠纷本站概不负责!如果侵犯了您的权益请与我们联系!
转载请注明出处: 免费源码网-免费的源码资源网站 » 蛋白质结构中pdb_strand_id和asym_id相互转化

发表评论 取消回复