MOE下载
别想了,要钱的。而且不算是主流软件,过。
Autodock和Autodock tools下载
下载地址:https://autodock.scripps.edu/download-autodock4/
如果你的电脑满足以下配置,那么推荐下载autodock GPU
- 操作系统:macOS Catalina 10.15.1 或更高版本。
- CPU:支持C++17的处理器
- 如Intel Core i5/i7或相应的AMD处理器。
- GPU:支持OpenCL的GPU,推荐使用:
- Apple / Intel Iris
- AMD Radeon(如Radeon Vega 64、Radeon VII)
- 其他现代AMD或NVIDIA GPU(如Radeon Pro系列)
- CUDA(可选):CUDA Toolkit 11或更高版本(适用于NVIDIA GPU)。
- 编译器:
- GCC 9或更高版本(支持C++17),可以通过Homebrew安装。
- Clang(macOS自带)也可以,但需要确认是否支持OpenMP。
- OpenCL:通常在macOS上自带。
下载完后我们得到autodock4和autogrid4两个程序文件。然后下载autodock tools
下载地址:https://ccsb.scripps.edu/mgltools/downloads/
推荐直接下载dmg文件,但是有可能无法通过验证从而无法安装,其次要注意的是:MGLTools 无法在 Catalina OS 下运行
(吗的,我就不能下载,公司电脑有安全性要求,不能下载)
PyMol下载
下载地址:https://www.pymol.org/
gromacs下载
brew install gromacs
下载成功后输入
gromacs --version
验证安装是否成功
PDB数据库使用
官方地址:https://www.rcsb.org/
首页如下:
我们以热休克蛋白HSP90AA1为例,其PDB ID为7DHG,所以我们在搜索栏输入7DHG:
主要关注红框里的几个地方。
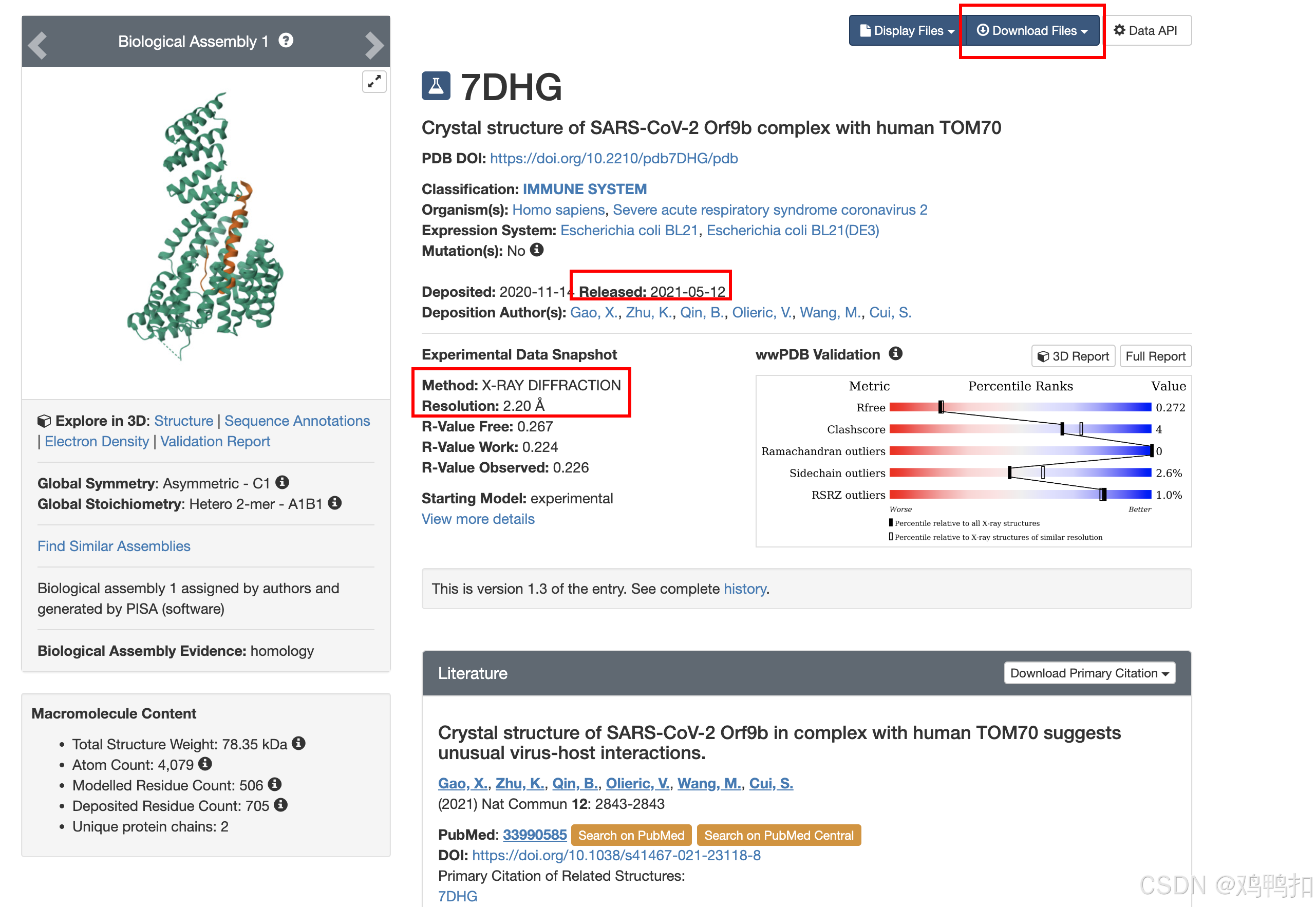
- Download 下载文件,一般选择
PDB Format即可 - Released 发表时间
- Method 一般只有X-Ray(X射线)和NMR两种。其中X射线最常见也最好
- Resolution 分辨率相关的指标,越小说明分辨率越高。一般小于2A就足够好了,具体看论文的指标
gromac能量最小化
运行 pdb2gmx
在终端进入包含你的PDB文件的目录,然后使用以下命令运行 pdb2gmx:
gmx pdb2gmx -f your_structure.pdb -o processed.gro -p topol.top -water spce
参数说明:
-f your_structure.pdb:输入的PDB文件。
-o processed.gro:输出的结构文件,通常为 .gro 格式。
-p topol.top:生成的拓扑文件名。
-water spce:指定使用的水模型(例如SPC/E水模型)。你可以根据需要选择不同的水模型。
选择力场
运行 pdb2gmx 后,系统会提示你选择一个力场(force field)。你会看到一个列表,选择适合你的模拟的力场(例如,CHARMM、AMBER、GROMOS等)。
力场列表如下:
1: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
3: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)
4: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
5: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006)
6: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)
7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
8: CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins)
9: GROMOS96 43a1 force field
10: GROMOS96 43a2 force field (improved alkane dihedrals)
11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
在这里我们选择5作为演示。输入5然后按enter。
处理缺失的原子
如果输入的PDB文件中有缺失的原子,pdb2gmx 可能会提示你处理这些缺失的原子。根据需要选择相应的选项。如下:
Fatal error:
Residue 165 named MET of a molecule in the input file was mapped
to an entry in the topology database, but the atom CG used in
that entry is not found in the input file. Perhaps your atom
and/or residue naming needs to be fixed.
gmx pdb2gmx 遇到了一个问题,提示你在输入文件中找不到某个残基(MET, 即甲硫氨酸)的特定原子(CG)。这个问题通常与PDB文件中的残基和原子命名不一致有关。
解决步骤
检查PDB文件:
打开 new_7dhg.pdb 文件,找到第165个残基(MET)。
检查这个残基的原子命名是否与GROMACS拓扑数据库中的命名一致。特别是查看是否有名为CG的原子。
修正残基命名:
如果原子CG缺失,可能是因为PDB文件中的命名不符合GROMACS的要求。你可以手动编辑PDB文件,确保所有原子都正确命名,或者添加缺失的原子。
你可以使用文本编辑器打开PDB文件,查找MET残基的定义(通常是以MET开头的行),并确保原子命名符合标准(如:N, CA, C, O, CB, CG等)。
使用其他工具检查结构:
你可以使用其他分子可视化工具(如PyMOL或Chimera)来查看和编辑PDB文件。这些工具可以帮助你更直观地发现问题。
重新运行 pdb2gmx:
修正完PDB文件后,保存更改,并重新运行 gmx pdb2gmx 命令。
本站资源均来自互联网,仅供研究学习,禁止违法使用和商用,产生法律纠纷本站概不负责!如果侵犯了您的权益请与我们联系!
转载请注明出处: 免费源码网-免费的源码资源网站 » 网络药理学:分子对接之一:macos上MOE和Autodock和PyMol和gromacs的下载、PDB数据库使用、gromacs能量最小化

发表评论 取消回复